Sequence alignment from the
phylogenetic
perspective
In phylogenetics, homology means..

Quiz: Why?
Structural alignment is another perspective
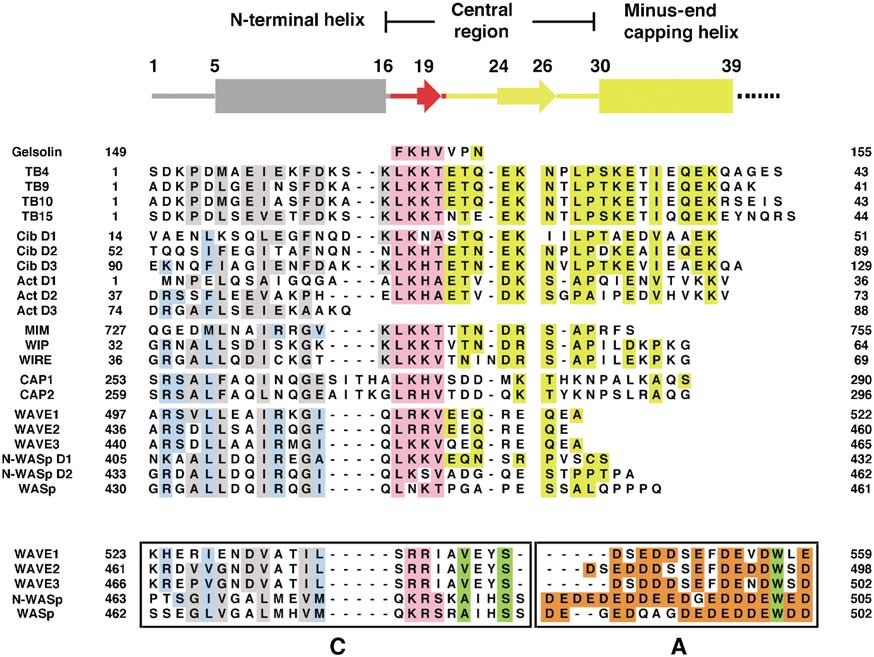
Sequence alignment for phylogenetics is hard!
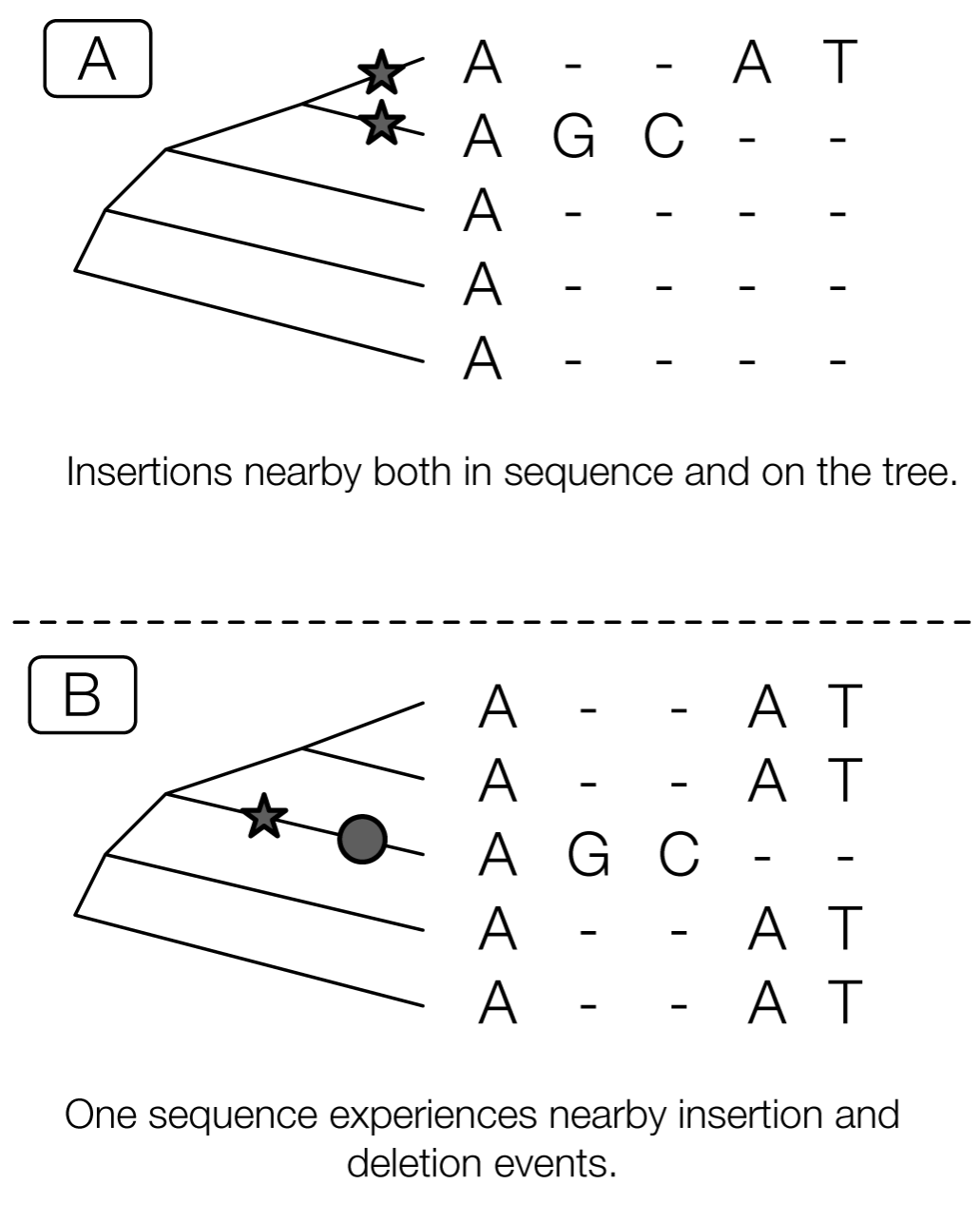
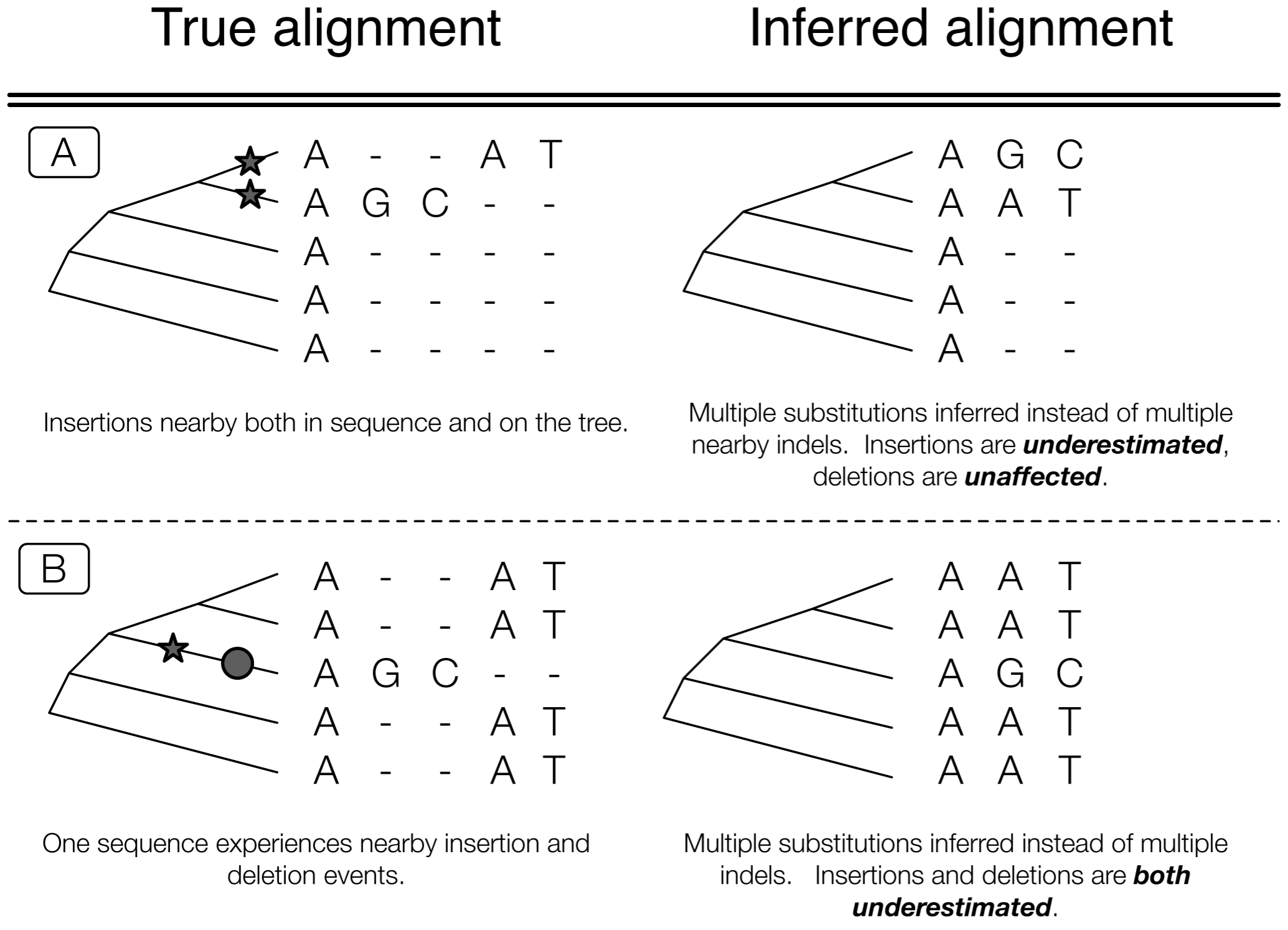
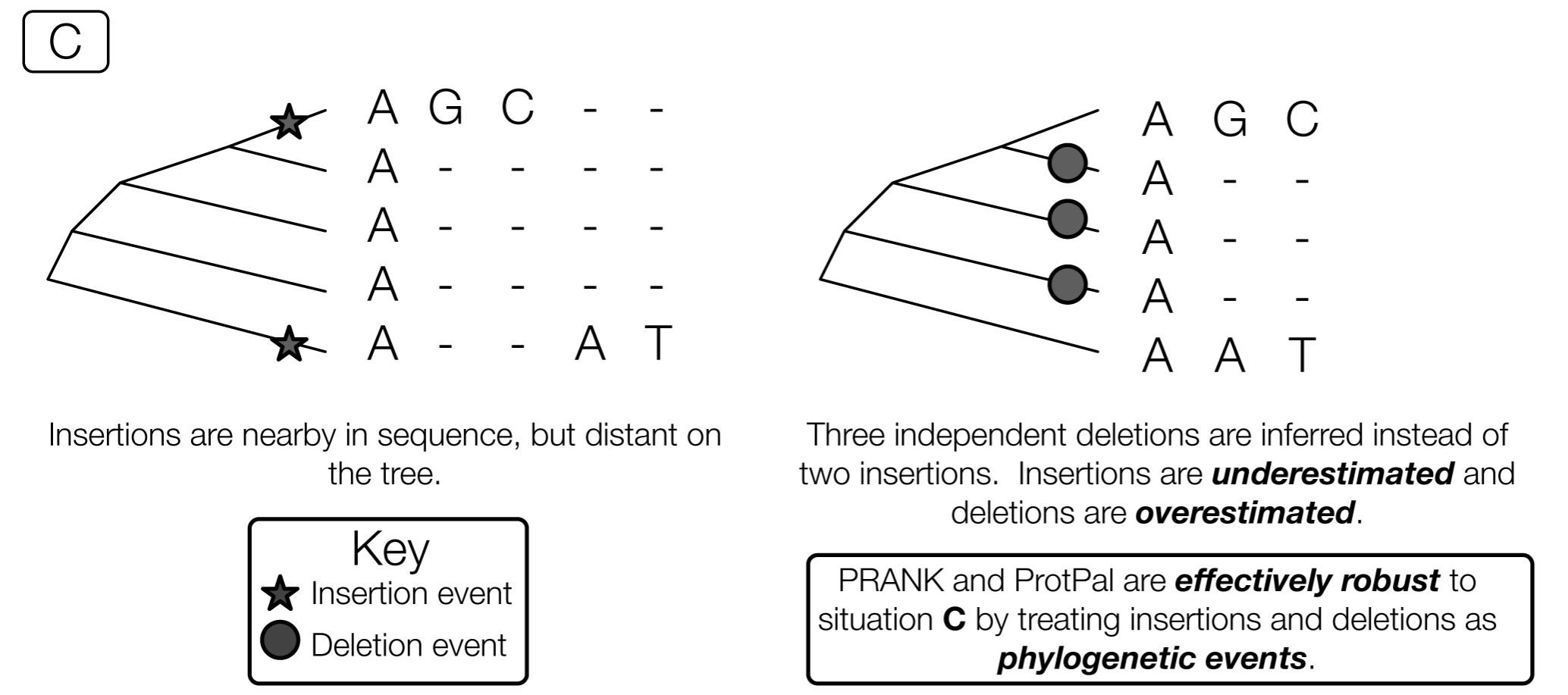
Sometimes it’s more or less impossible
Phylogenetic approach is best
Clustal W vs PRANK
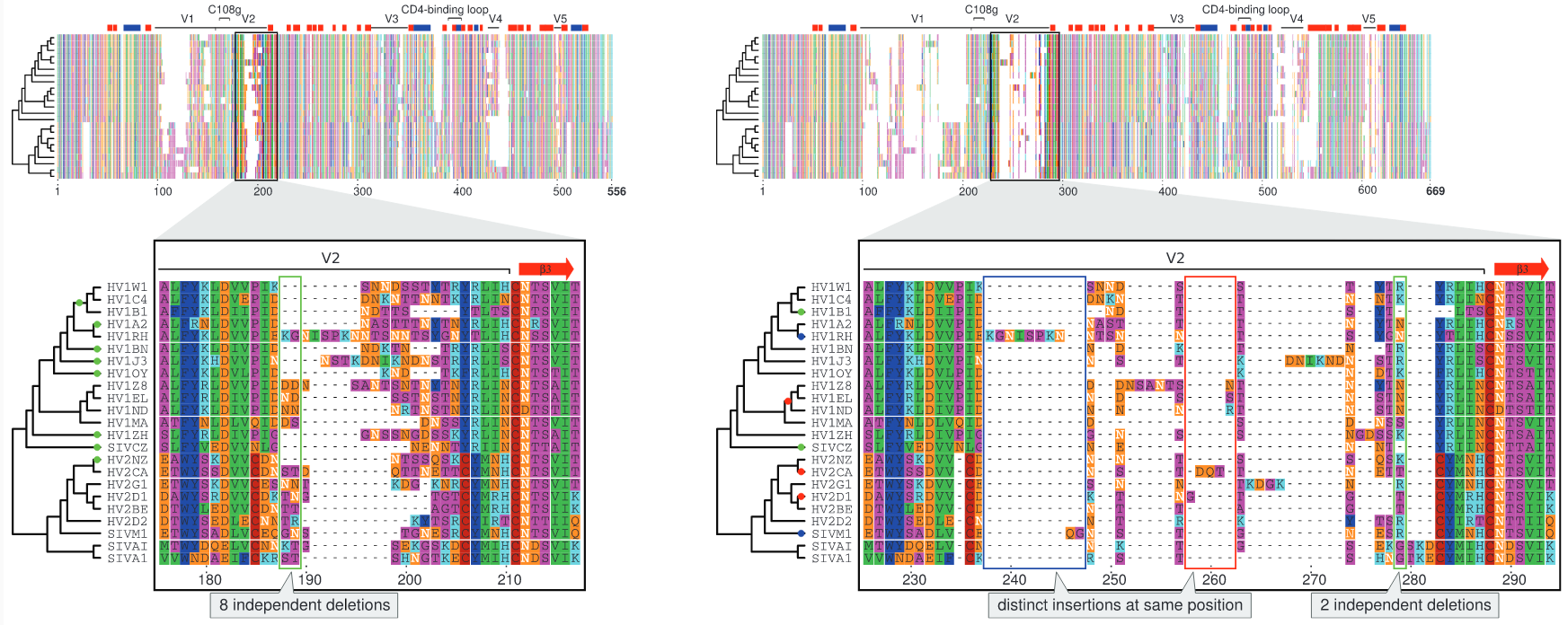
PRANK is a smart “hack”
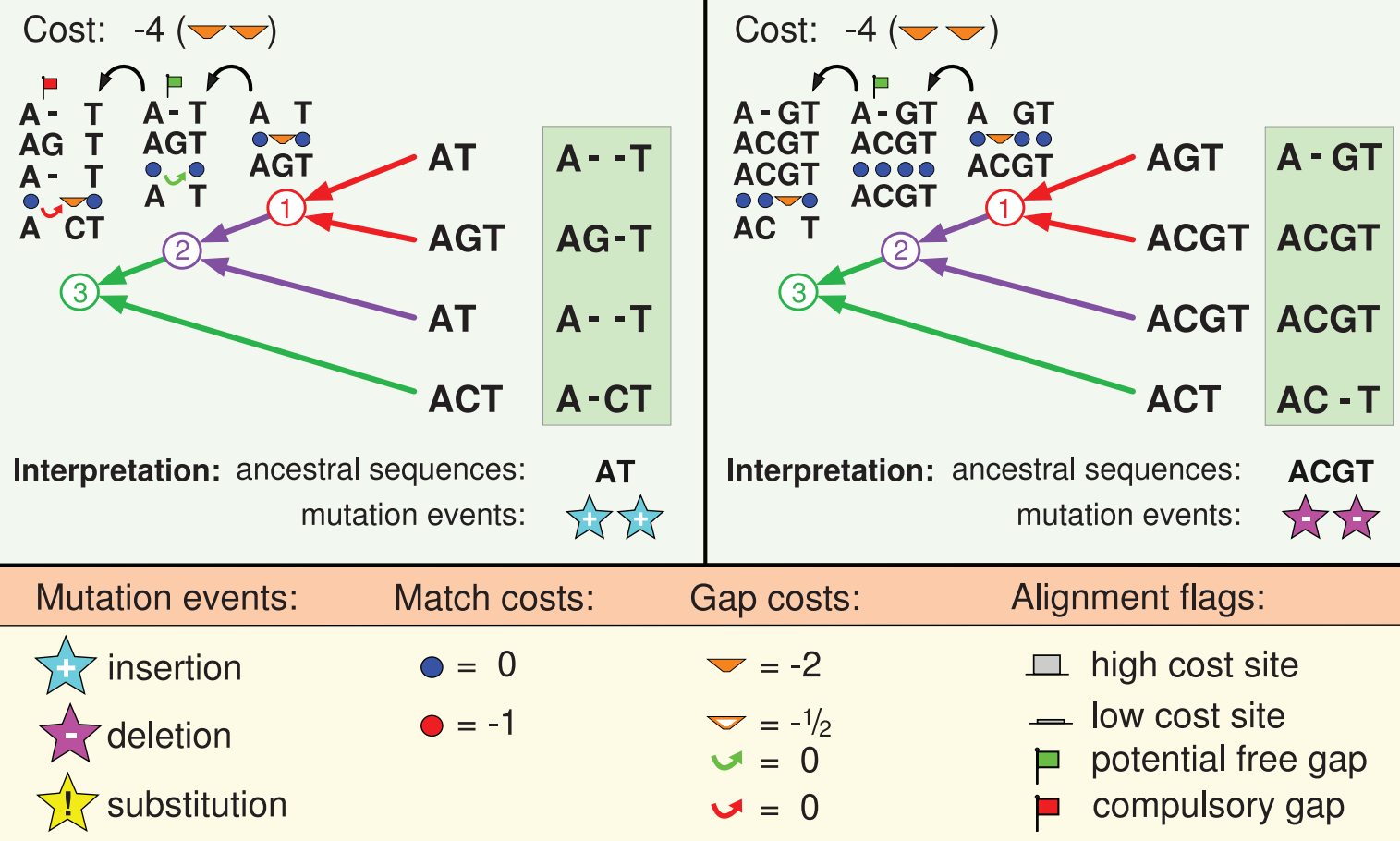
BAli-Phy is fully model-based

BAli-Phy is fully model-based

BAli-Phy is fully model-based

BAli-Phy is fully model-based

BAli-Phy is fully model-based
Note how BAli-Phy can trace the history of each site through
the
tree, and the columns are simply a reflection of that.
BAli-Phy estimates alignment uncertainty
BAli-Phy is fully Bayesian, so it has a rigorous notion of alignment uncertainty, which can be elegantly summarized by that software:

What’s funny about this protein-coding alignment?

What’s funny about this protein-coding alignment?
