Mutation and selection in B cells
Thanks to Sarah Cobey for some of the slides
Structure of a B cell receptor
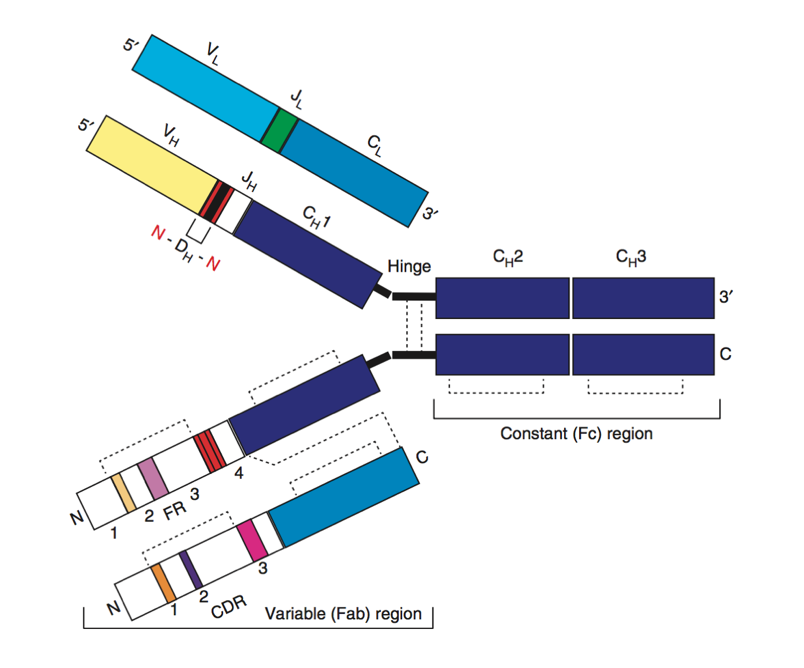
VDJ recombination

modified from Murugan et al. 2012
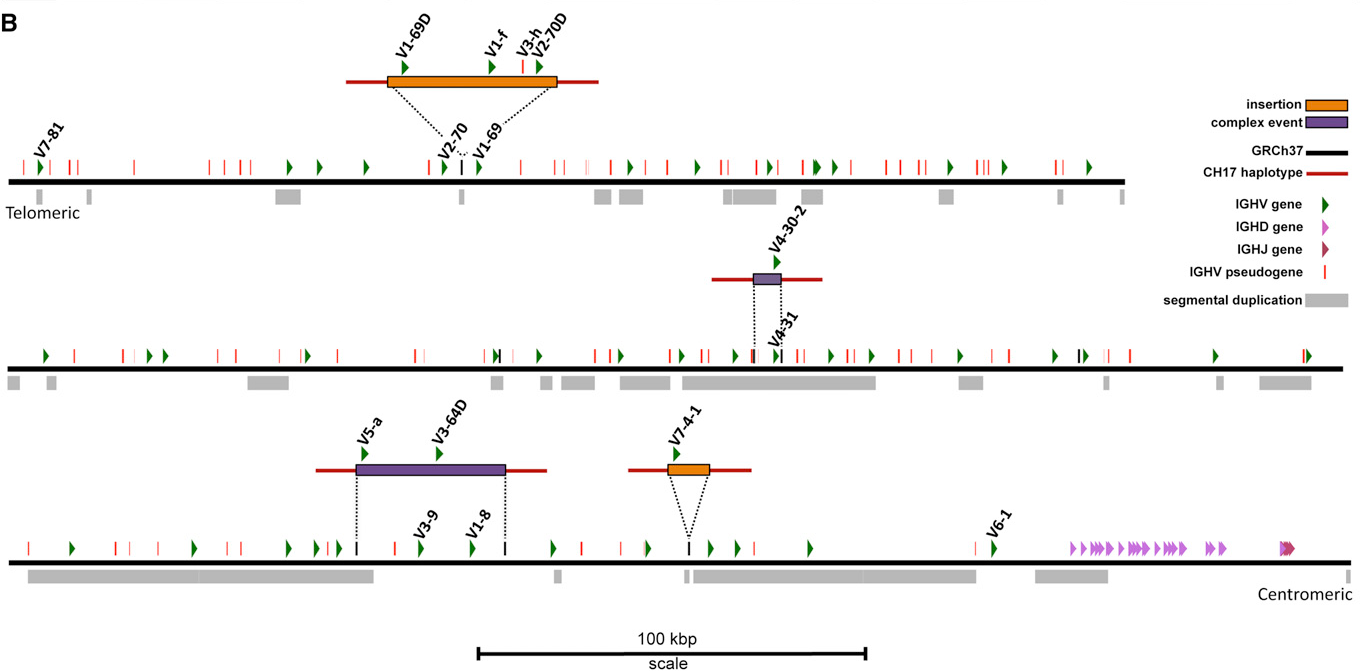
VDJ loci










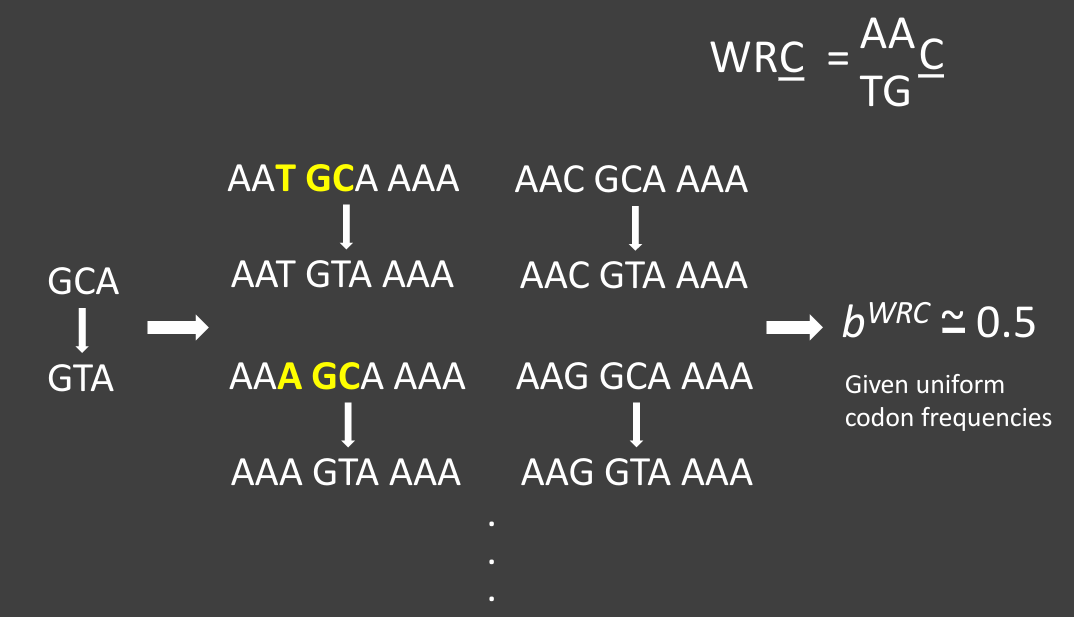
Somatic hypermutation is AID + repair

The mutations have peculiar biases

The mutations have peculiar biases

Phylogenetics is not straightforward
Incorporating context sensitivity helps performance

Natural selection inferences are also not straightforward

Natural selection inferences are also not straightforward

Natural selection inferences are also not straightforward
Context-sensitive mutation can confound
natural selection
inference for B cells.
Productive vs. out-of-frame receptors

Out-of-frame reads can be used to infer neutral mutation rate.
One would like to quantify these biases

Classic work by Kleinstein group
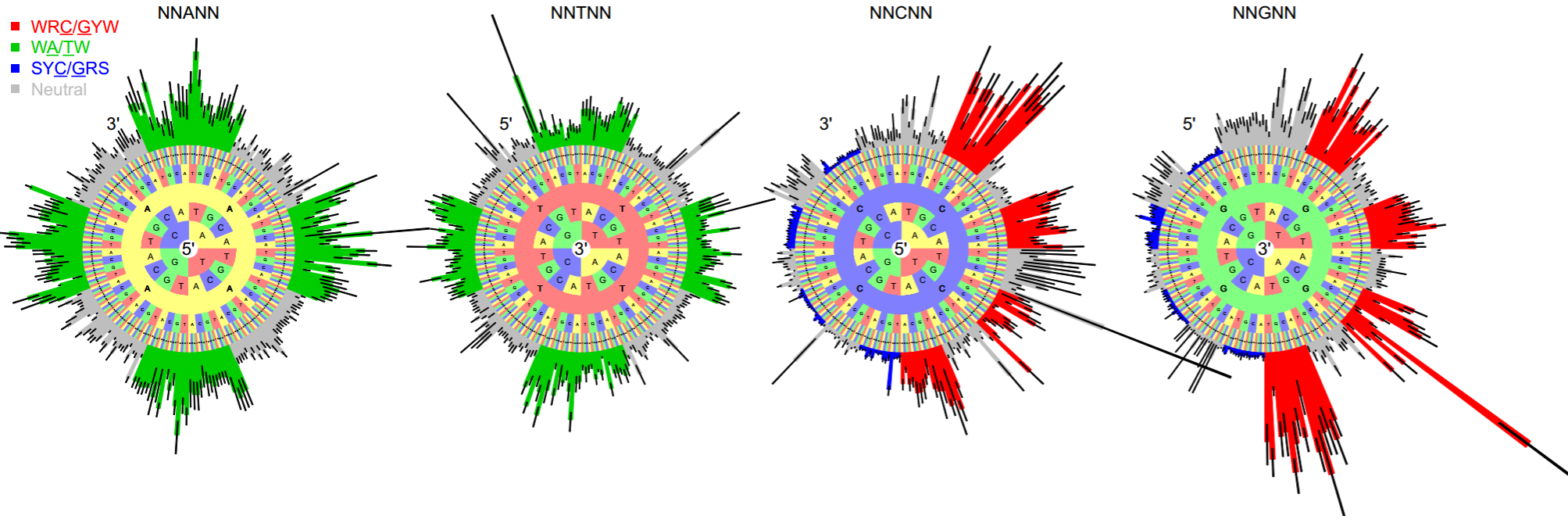
- Yaari, Vander Heiden, Uduman, Gadala-Maria, Gupta, Stern, Kleinstein, Front Immunol, 2013.
- Cui, Di Niro, Vander Heiden, Briggs, … , Kleinstein J Immunol, 2016.
Have extended this framework

We can add overlapping motifs.
Now the \(\theta\) entry for a 5-mer
answers the question:
what is this 5-mer telling me that the inner
3-mer did not?
These get automatically zeroed out if not informative.

Feng, Shaw, Minin, Simon, & M, arXiv, in revision for Ann. Applied Stat.
Motivation



A simple infinite-type branching process model
- \(p\): probability of division
- \(q\): probability of mutation

“GCTree” likelihood



GCtree works in simulation

GCtree finds more common IgH and IgL trees
